
良种繁育
基因组预测的序列数据在家畜育种中的应用
|
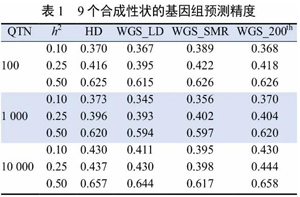
表1 列出了9个合成性状的基因组预测精度。当QTN的数量较小时,可以识别能支撑该性状的遗传变异的变体(variants)具有足够的统计功效(statistical power),使用这些变体(WGS_SMR)进行预测的准确性高于用来自商业标记阵列(HD)的标记进行预测的。这与之前的观察结果一致,添加一个或几个具有较大作用的标记作为预测因子可以提高该标记序列的预测精度。 当QTN的数量较大时,WGS_SMR的性能比HD的差。在这种情况下,从序列数据中选择的其他变异集可能(略微)比商业标记序列更有利,因为它们不会像商业标记序列那样受到确定偏倚(ascertainment bias)的影响。 这些结果部分是由于目前使用商业标记阵列进行基因组选择已经获得了很高的预测准确性,且与其他研究结果一致。后者发现,与HD标记阵列相比,序列数据在基因组预测上没有改善或只有微小的变化。有待确定的是,结果是否会因以下原因而得到改善:来自多个品种的数据,使用多品种测试和更大的测试集,或比岭回归更适合于大规模开发序列数据的基因组预测方法。 3 结论 无论种群的规模多大,只要个体与具有标记阵列或序列数据的亲缘联系在一起,同时该亲缘有足够多的信息,恰当的测序策略和“杂交剥离”的结合是在大群的纯种系谱中生成全基因组序列数据的一种有效方法。 目前尚不清楚,这些带有估测序列数据的大数据集是否能够提高基因组预测的准确性。 |
上一篇:宠物兔如何引进种公种母?
下一篇:青海选育二十载牦牛有了新品种